MQLNKV-L(U13C6,15N)-KGL(U13C6,15N)MIALPVMAIAA-dipalmitoyl2C-SSNKNGG-K-biotin, which upon cleavage by LspA, yields the product peptide dipalmitoyl2C-SSNKNGG-K-biotin. A product standard [dipalmitoyl2C-SSNKNAAK-(NHCH2CH2NH)-biotin; CPC Scientific] was in the reaction mixture as an internal standard for normalization of product quantitation.
Abstract
Clinical development of antibiotics with novel mechanisms of action to kill pathogenic bacteria is challenging, in part, due to the inevitable emergence of resistance. A phenomenon of potential clinical importance that is broadly overlooked in preclinical development is heteroresistance, an often-unstable phenotype in which subpopulations of bacterial cells show decreased antibiotic susceptibility relative to the dominant population. Here, we describe a new globomycin analog, G0790, with potent activity against the Escherichia coli type II signal peptidase LspA and uncover two novel resistance mechanisms to G0790 in the clinical uropathogenic E. coli strain CFT073. Building on the previous finding that complete deletion of Lpp, the major Gram-negative outer membrane lipoprotein, leads to globomycin resistance, we also find that an unexpectedly modest decrease in Lpp levels mediated by insertion-based disruption of regulatory elements is sufficient to confer G0790 resistance and increase sensitivity to serum killing. In addition, we describe a heteroresistance phenotype mediated by genomic amplifications of lspA that result in increased LspA levels sufficient to overcome inhibition by G0790 in culture. These genomic amplifications are highly unstable and are lost after as few as two subcultures in the absence of G0790, which places amplification-containing resistant strains at high risk of being misclassified as susceptible by routine antimicrobial susceptibility testing. In summary, our study uncovers two vastly different mechanisms of resistance to LspA inhibitors in E. coli and emphasizes the importance of considering the potential impact of unstable and heterogenous phenotypes when developing antibiotics for clinical use.
SOCIAL MEDIA
Connect with us and stay updated by following our social media channels.
Latest Briefings from our Knowledge Center
Press Releases, Industry News, Articles, and Technical Content

Home / Knowledge Center / Articles
Highlights from CPC Scientific’s 2025 Annual Conference
GMP Peptide Manufacturing
We’re excited to share the key moments from CPC Scientific’s 2025 Annual Conference!
Hosted in San Jose, California, our annual gathering brought together colleagues from various teams and timezones for meaningful updates, focused […]

At NextGen Biomed 2025, Joseph Denby delivered a cutting-edge presentation highlighting the critical role green chemistry plays in the sustainable production of peptide APIs and peptide–oligo conjugates...

Home / Knowledge Center / Articles
CPC Scientific at DCAT 2025!
GMP Peptide Manufacturing
What an amazing time we had at DCAT 2025! It was fantastic to meet with colleagues, clients, and friends, and showcase CPC Scientific’s cutting-edge capabilities in synthetic API development and commercial production.
A heartfelt […]

We are excited to share that CPC Scientific Inc. has officially received the Good Manufacturing Practice (GMP) Certificate in accordance with the ISO 22716:2007(E) Cosmetic Guidelines.
This certification highlights our unwavering commitment to delivering the highest quality peptide ingredients for cosmetic use, guaranteeing:
- Superior Quality Control – Striving for excellence in safety, consistency, and purity.
- Global […]

In Part 1 of our Minimal Protection Group Strategies for SPPS, we discussed methods for eliminating sidechain protection on hydroxy-bearing amino acids such as serine, threonine, tyrosine, and hydroxyproline. By omitting t-butyl protection, we enhanced atom economy and avoided the use of hazardous solvents typically required to remove these protection groups. In Part 2, we present a new case study, expanding our approach to include the unprotected side chains of histidine, tryptophan, and arginine. We demonstrate the synthesis of a Goserelin peptide API impurity, showcasing how a convergent peptide fragment strategy can be used to eliminate the need for TFA and diethyl ether, eliminate side chain protection of Arginine, Histidine, and Tryptophan.
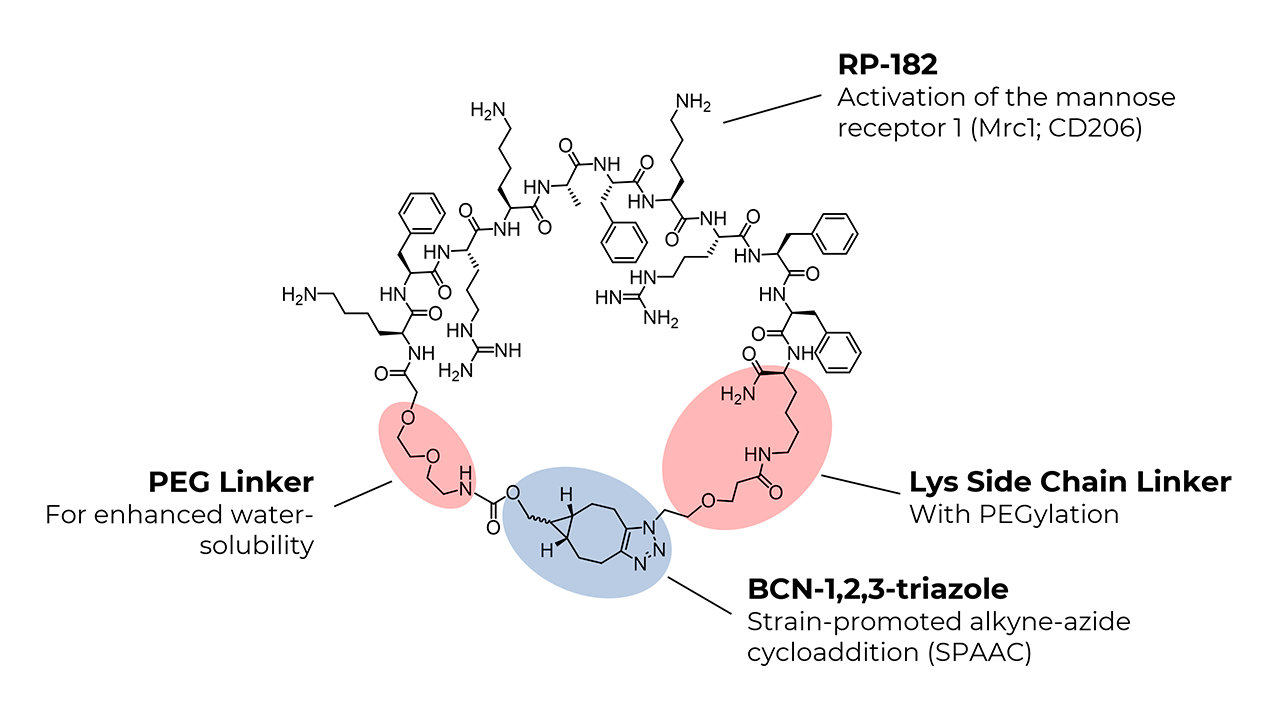
The synthesis of the linear RP-182 analog, bicyclo[6.1.0]non-4-yn-9-ylmethyloxycarbonyl-PEG2-Lys-Phe-Arg-Lys-Ala-Phe-Lys-Arg-Phe-Phe-Lys(azido-PEG)-NH2, was achieved using standard solid-phase peptide synthesis (SPPS) protocols. After cleaving the linear peptide from the resin, macrocyclization was performed in the liquid phase through a strain-promoted click reaction. BCN introduces extra ring strain due to its fused cyclopropane structure. The combined effect of ring strain, the selection of BCN, and copper catalysis significantly increases the macrocyclization efficiency of longer peptides like RP-182.
Nakagawa, Mayumi, Teresa Evans, Milan Bimali, Hannah Coleman, Jasmine Crane, Nadia Darwish, Jennifer L. Faulkner et al. MedRxiv (2025): 2025-01.
The vaccine consisted of four current good manufacturing production-grade synthetic peptides covering the HPV 16 E6 protein [amino acid (aa)1-45, 46-80, 81-115, and 116-158] (CPC Scientific, San Jose, CA [..]

Stapled peptides have emerged as a powerful tool in drug discovery and therapeutic development due to their ability to overcome the limitations associated with traditional peptide drugs, such as poor stability and low cell permeability. By introducing staples into the peptide backbone, researchers can stabilize peptide conformations and enhance their interactions with target proteins, resulting in improved efficacy and specificity. This approach not only addresses the challenges of peptide drug design but also opens new avenues for targeting challenging biomolecular interactions that are difficult to modulate with small molecules or antibodies. The development of stapled peptides has led to significant advancements in targeting protein-protein interactions, addressing previously intractable diseases, and enhancing the precision of therapeutic interventions.
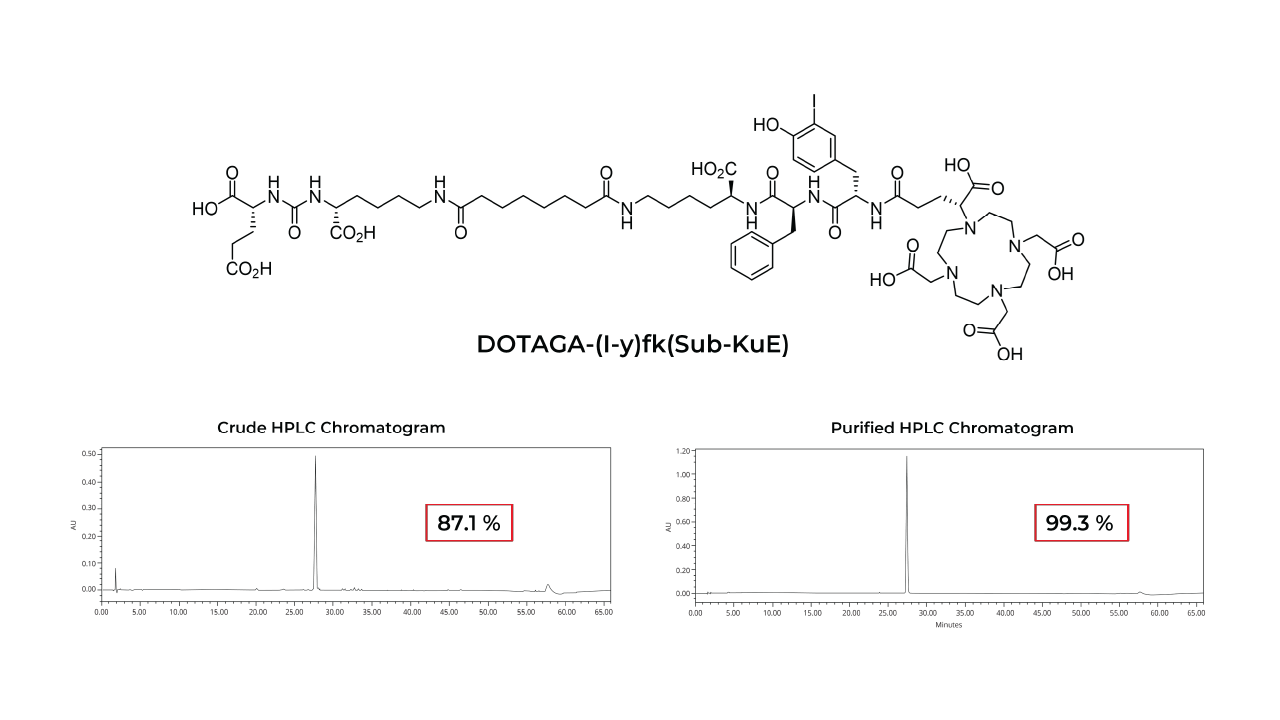
Peptide receptor radionuclide therapy (PRRT) is a targeted therapeutic approach that utilizes peptides to deliver cytotoxic radiation to specific receptors overexpressed on cancer cells. Peptides offer several advantages as therapeutic vectors in PRRT due to their small size, favorable pharmacokinetics, high binding affinity, low immunogenicity and toxicity, and minimal off-target effects. Tumor-targeting peptides conjugated to radionuclide chelates represent a promising class of cancer therapeutics.