CPC SCIENTIFIC WHITE PAPERS
Recent peptide- and oligonucleotide-based articles designed to increase your understanding about various subject matters.
Delivery of polypeptides in multi-kilogram quantities within commercially competitive timelines is extremely challenging, especially when coupled with a desire to minimize environmental and economic impact. This case study explores how CPC Scientific’s advanced process improvements and efficiencies within solid-phase peptide synthesis (SPPS) have enabled multi-kilogram delivery of a pharmaceutically-relevant decapeptide within a challenging timescale, driving sustainable and cost-saving production for the client.

Our team has developed an innovative DMF recycling strategy that substantially reduces solvent consumption during solid-phase peptide synthesis. Minimizing use of DMF, a major environmental and cost contributor in peptide manufacturing, has improved process sustainability and cost efficiency. This method neatly demonstrates how targeted green chemistry practices can be successfully integrated into large-scale SPPS, supporting more environmentally responsible and economically viable peptide production.

In Part 1 of our Minimal Protection Group Strategies for SPPS, we discussed methods for eliminating sidechain protection on hydroxy-bearing amino acids such as serine, threonine, tyrosine, and hydroxyproline. By omitting t-butyl protection, we enhanced atom economy and avoided the use of hazardous solvents typically required to remove these protection groups. In Part 2, we present a new case study, expanding our approach to include the unprotected side chains of histidine, tryptophan, and arginine. We demonstrate the synthesis of a Goserelin peptide API impurity, showcasing how a convergent peptide fragment strategy can be used to eliminate the need for TFA and diethyl ether, eliminate side chain protection of Arginine, Histidine, and Tryptophan.
The synthesis of the linear RP-182 analog, bicyclo[6.1.0]non-4-yn-9-ylmethyloxycarbonyl-PEG2-Lys-Phe-Arg-Lys-Ala-Phe-Lys-Arg-Phe-Phe-Lys(azido-PEG)-NH2, was achieved using standard solid-phase peptide synthesis (SPPS) protocols. After cleaving the linear peptide from the resin, macrocyclization was performed in the liquid phase through a strain-promoted click reaction. BCN introduces extra ring strain due to its fused cyclopropane structure. The combined effect of ring strain, the selection of BCN, and copper catalysis significantly increases the macrocyclization efficiency of longer peptides like RP-182.

Stapled peptides have emerged as a powerful tool in drug discovery and therapeutic development due to their ability to overcome the limitations associated with traditional peptide drugs, such as poor stability and low cell permeability. By introducing staples into the peptide backbone, researchers can stabilize peptide conformations and enhance their interactions with target proteins, resulting in improved efficacy and specificity. This approach not only addresses the challenges of peptide drug design but also opens new avenues for targeting challenging biomolecular interactions that are difficult to modulate with small molecules or antibodies. The development of stapled peptides has led to significant advancements in targeting protein-protein interactions, addressing previously intractable diseases, and enhancing the precision of therapeutic interventions.
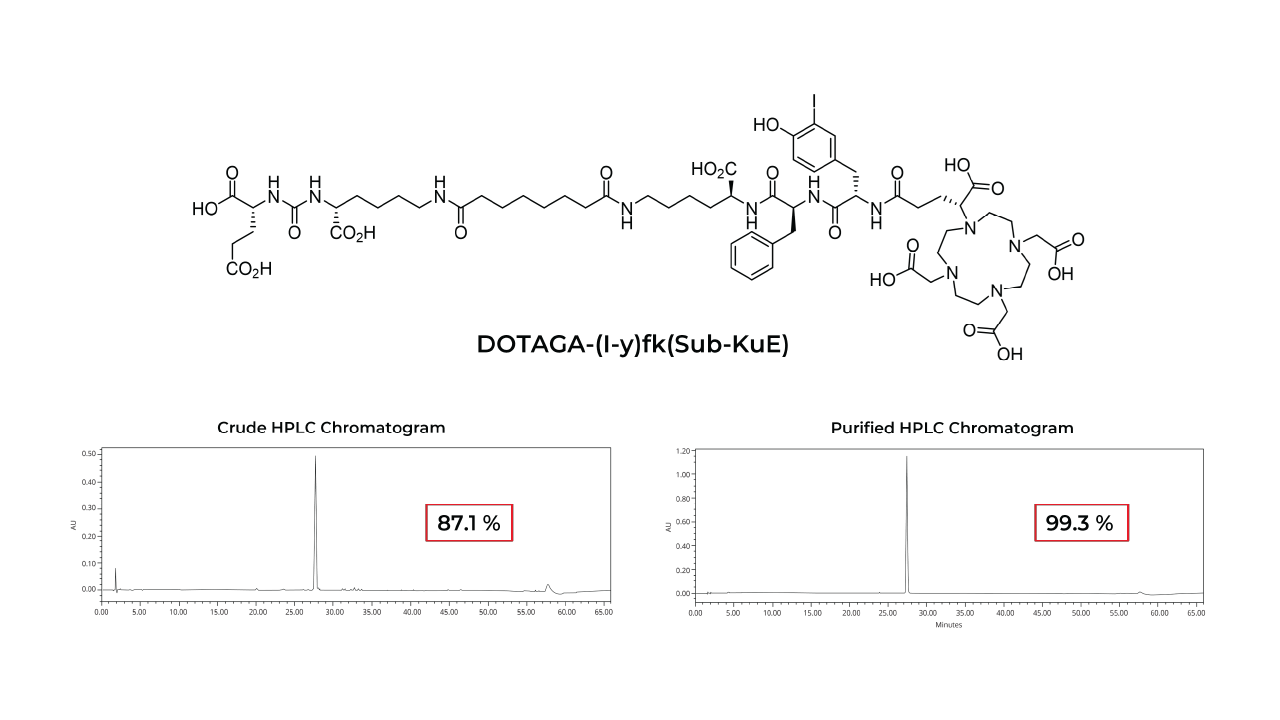
Peptide receptor radionuclide therapy (PRRT) is a targeted therapeutic approach that utilizes peptides to deliver cytotoxic radiation to specific receptors overexpressed on cancer cells. Peptides offer several advantages as therapeutic vectors in PRRT due to their small size, favorable pharmacokinetics, high binding affinity, low immunogenicity and toxicity, and minimal off-target effects. Tumor-targeting peptides conjugated to radionuclide chelates represent a promising class of cancer therapeutics.
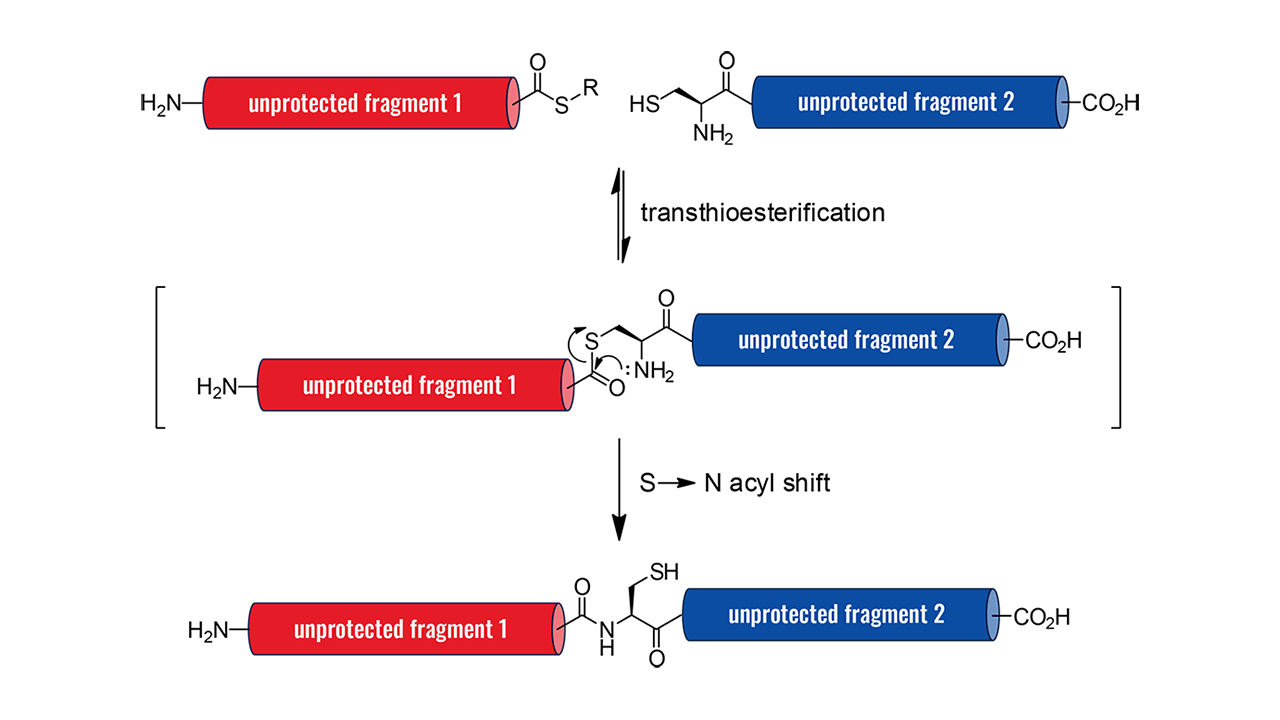
Most proteinogenic peptides, except for certain hydrophobic sequences, can be synthesized in a linear fashion using solid-phase peptide synthesis (SPPS) methods. However, longer sequences, particularly those exceeding 70 amino acids, often require alternative techniques due to challenges like steric hindrance. Other issues, such as poor solvation of the protected peptide during synthesis and the formation of intermolecular hydrogen bonds (e.g., β-sheets) between fragments, can also result in inefficient coupling and deprotection.

Synthetic oligonucleotides constitute an important class of therapeutics developed to treat a variety of indications. Two main synthetic approaches exist for the conjugation of a peptide to an oligonucleotide: parallel and linear. The primary benefit of the linear approach is the one-pot solid-phase assembly and compatibility with machine automation. However, in cases where poor compatibility of peptide and oligo chemistries exist or long peptide and oligo fragments are required, preparing both components separately and linking both compounds together may offer the simplest solution.

Solid-phase peptide synthesis (SPPS) approaches require that the side chains of certain amino acids be protected from undesired reactivity during synthesis. The installation and removal of these protection groups results in a lower atom economy in the production process. Removal of the protection groups often requires large volumes of trifluoroacetic acid (TFA) or other strong acids which can result in lower yields and pose a significant risk to the environment.
The transferred energy from a fluorescent donor is converted into molecular vibrations if the acceptor is a non-fluorescent dye (quencher). When the FRET is terminated (by separating donor and acceptor), an increase of donor fluorescence can be detected. The design and synthesis work at CPC for FRET and TR-FRET peptide substrates include modification of sequences, selection of donor/quencher pairs, improvement of FRET substrate solubility and quenching efficiency.
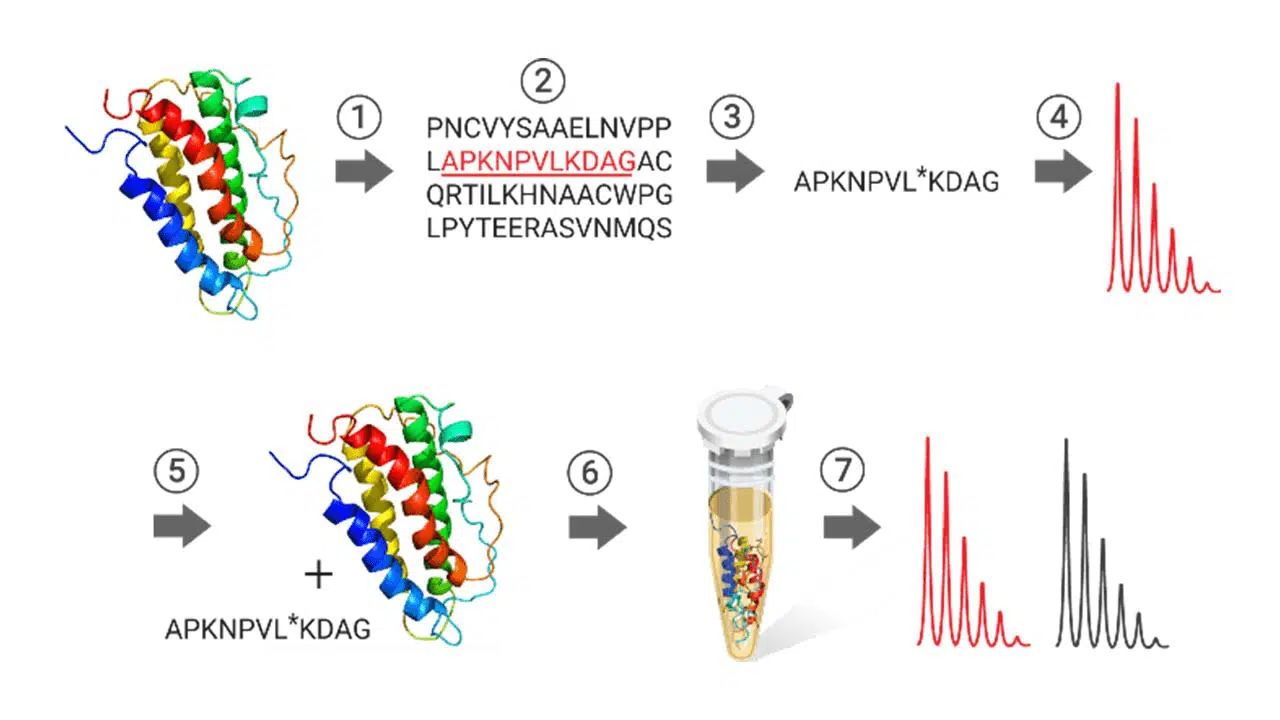
Genomics research shows that more than a million proteins are encoded by approximately 30,000 human genes. Proteomics, the study of proteins encoded by the genome, includes identifying post-translational modifications, structural analyses, protein localization studies, and protein quantitation. Mass spectroscopybased techniques have evolved as a powerful tool in proteomics. Stable isotope-labeled peptides (SIL peptides) are chemically and physically indistinguishable from their endogenous counterparts concerning retention time, ionization efficiency, and fragmentation pathways.
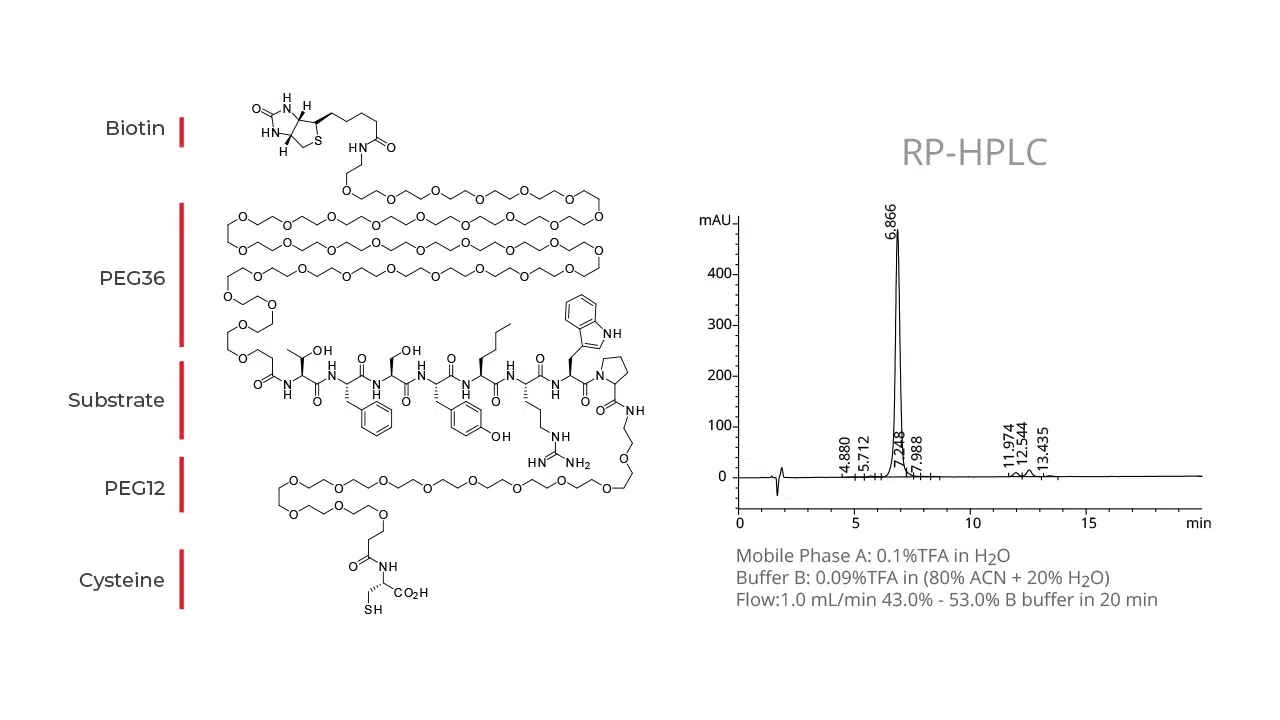
Peptides play a vital role in the pharmaceutical industry and drug therapeutic development; however, their in vivo applications are sometimes limited due to fast degradation by proteases, poor solubility, antigenic responses, and glomerular filtration in the kidney. The covalent attachment of polyethylene glycol (PEG) chains to peptides is one approach that can reduce immunogenicity, improve solubility, and reduce renal clearance.
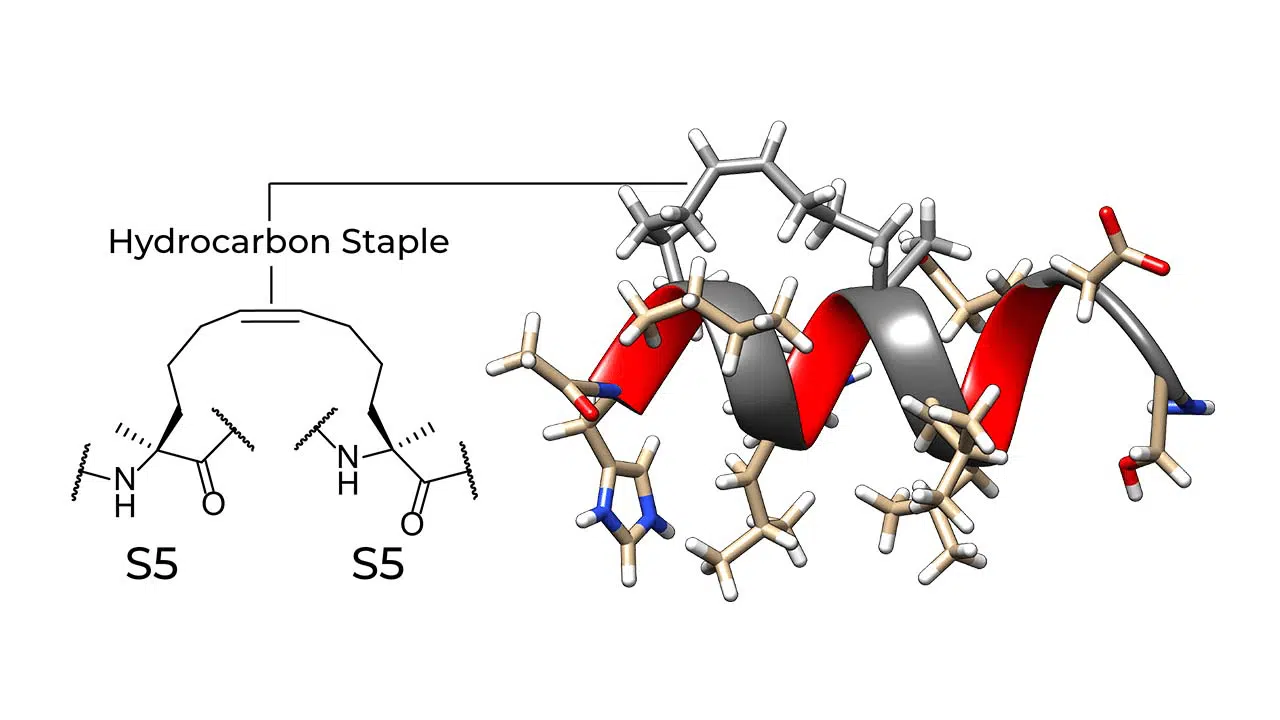
Hydrocarbon-stapled peptides are locked into their bioactive alpha-helical conformation through the site-specific introduction of a chemical brace, an all-hydrocarbon staple. The idea of peptide stapling was introduced to overcome the limitations of two broad classes of therapeutic agents (small molecules and protein biologics) in targeting intracellular protein-protein interactions. Small molecules only work on proteins with a specific surface feature, and most protein biologics do not penetrate cells. Because stapled peptides are locked into a stabilized α-helical structure (the most common element of protein secondary structures), they can easily penetrate cells.
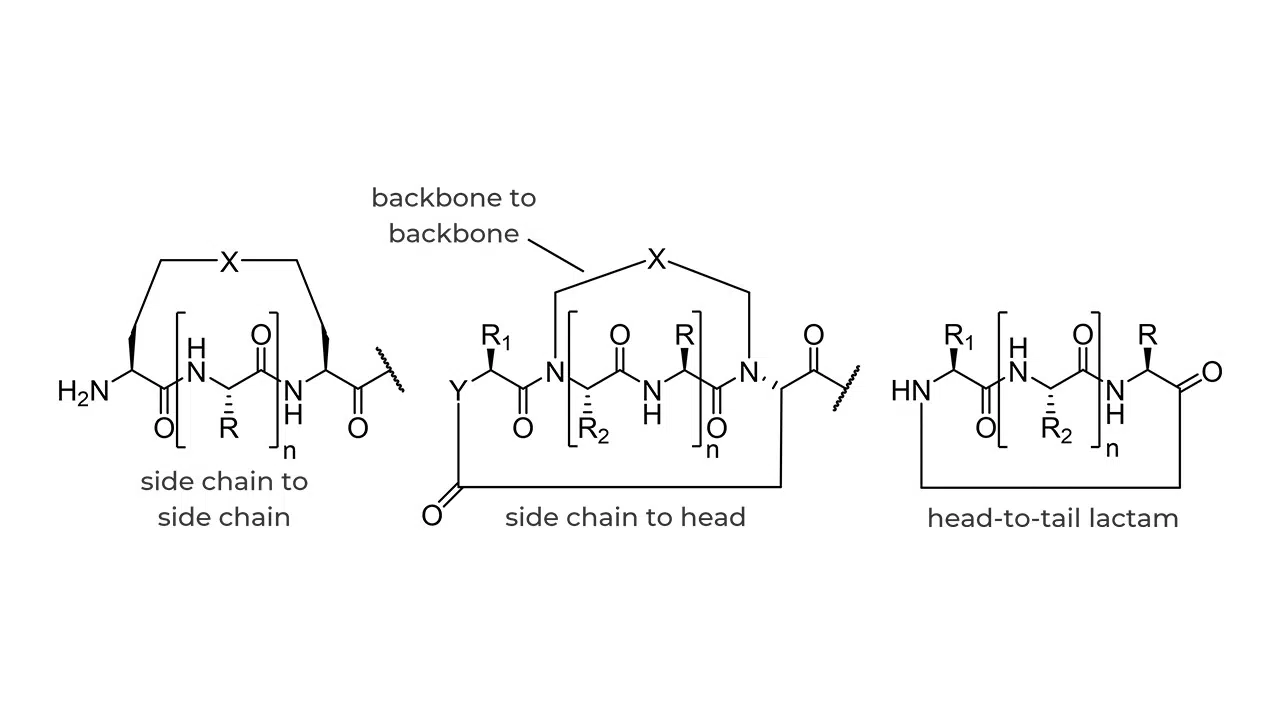
Protein-protein and protein-peptide interactions play critical roles in all types of cellular processing. Peptides are natural partners to proteins and, as ligands, bind to proteins with high affinity due to their capacity to adapt to the often flexible protein surface. Despite this, peptides have drawbacks as drug candidates that include low plasma bioavailability, instability from proteolytic enzymes, and poor passive membrane permeability. Some success has been achieved with linear peptides, particularly peptides that maintain α-helical secondary structures. These motifs can be introduced to stabilized α-helical motifs by common “peptide-stapling” approaches, but stapled peptides can suffer from low bioactivity and poor solubility. Another strategy to maintain peptide secondary structure is modification by macrocyclization.
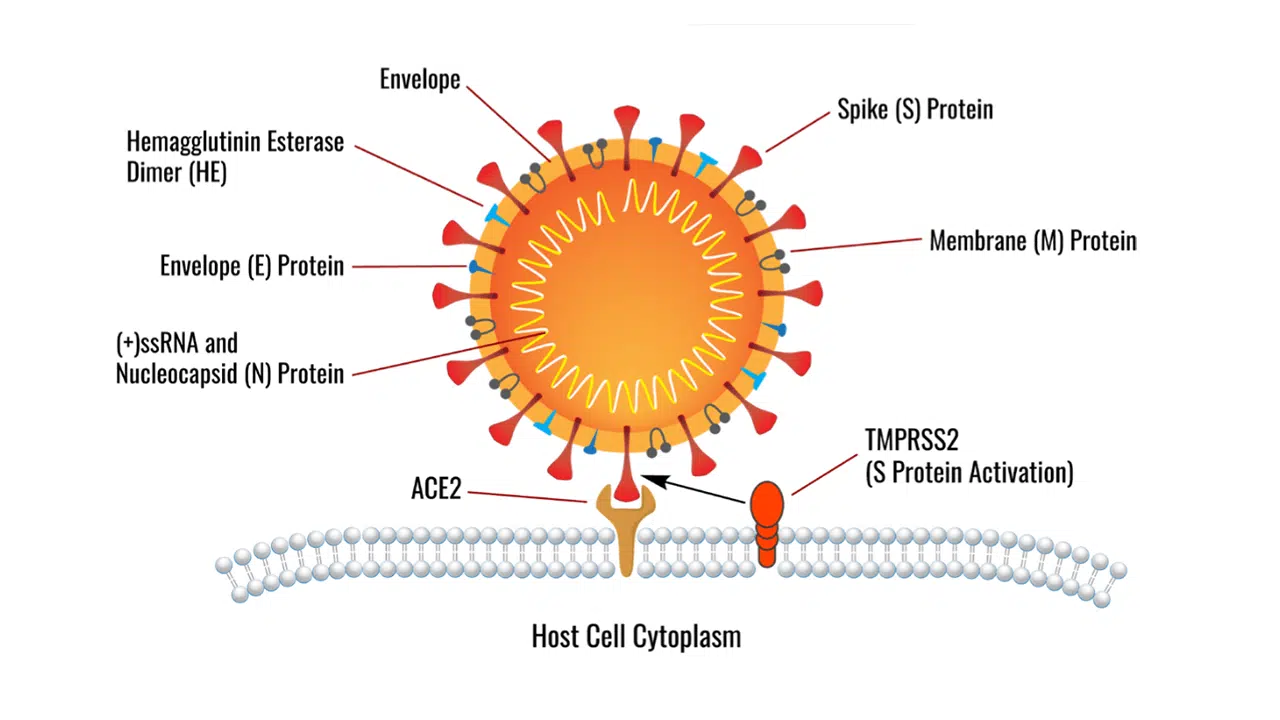
Mutations to the RBD of SARS-CoV have resulted in the reorganization of SARS-CoV-2 RBD, specifically to the loops that are in close proximity to the ACE2-binding ridge. Due to these structural changes, an additional intramolecular main-chain hydrogen bond is gained between Asn487 and Ala475 (Figure 4a). The Asn487–Ala475 hydrogen bond causes the RBM of SARS-CoV-2 to be more structurally compact, enabling the loop containing Ala475 to move into closer proximity to the ACE2 α1 helix. Closer contact between the RBM and α1 helix of ACE2 results in more intermolecular interactions (Figure 4a,b). Some of these interactions include hydrogen bonds between Gln493 (SARS-CoV-2) and Glu35 (ACE2),[3b,c] and main-chain hydrogen bonds between Gly502 and Lys31.
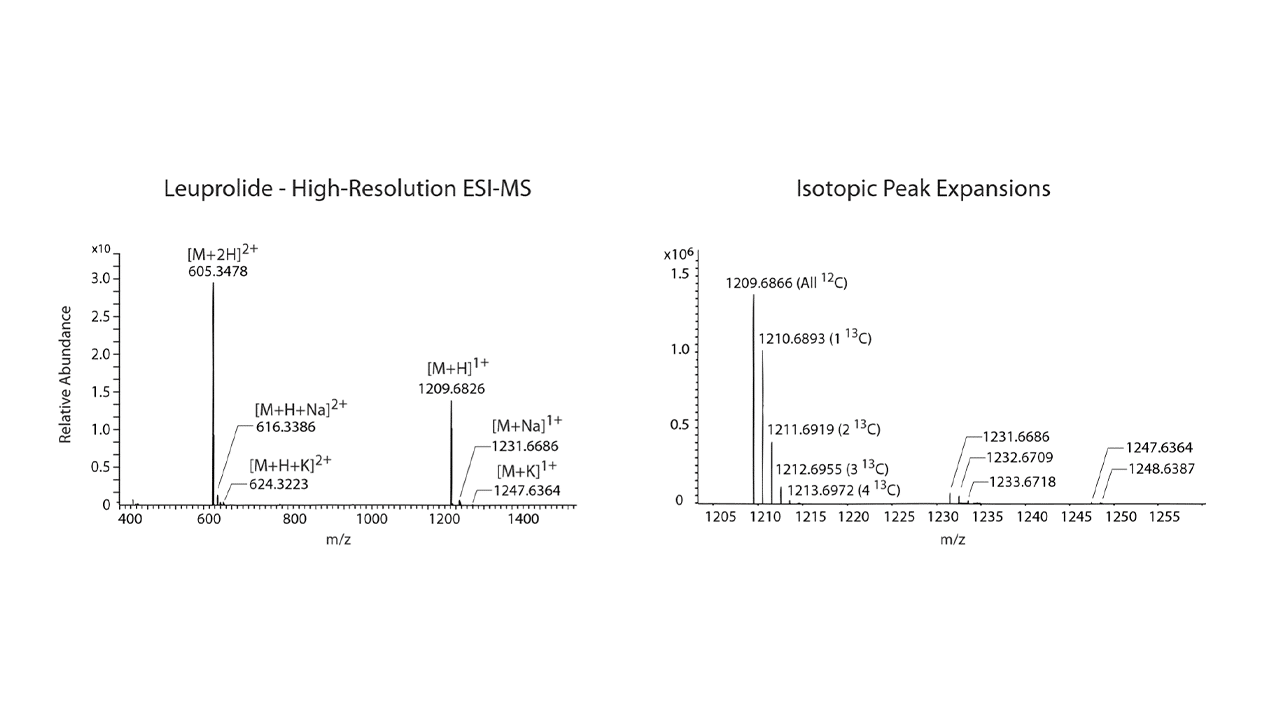
This guide will help you understand how to read ESI spectra for large and complex bio-molecules like peptides. Other techniques such as high-performance liquid chromatography (HPLC) show one major peak (i.e., absorbance and purity for RP-HPLC) in the spectra, making the interpretation straightforward. ESI-MS spectra are more complicated due to the multiplicity of the data. While most small molecules (100-500 AMU) behave well in ESI-MS and ionize as a single charged state, larger biomolecules, such as peptides, ionize as multiple charged variants. A major advantage of ESI-MS over other mass spectroscopy techniques is that molecular fragmentation is rare (t-Boc groups and a few other protection groups are an exception), which is why ESI-MS is often referred to as a soft ionization technique.

Solid-phase peptide synthesis (SPPS) has many advantages over liquid-phase peptide synthesis (LPPS) for preparing and manufacturing synthetic peptides. Except the synthesis of short peptide sequences (i.e., less than five amino acid residues), SPPS is faster, more efficient, and more economical than liquid-phase peptide synthesis (LPPS). Some of the advantages of SPPS include: (1) Excess reagents and products can be easily washed away, (2) using excess reagents to increase reaction rates and drive reactions to completion, (3) intermediates do not require isolation or characterization, (4) access to a broader range of solvents with low volatility and high polarity, (5) tethered peptide provides a ‘pseudo-dilute’ microenvironment, which can inhibit intermolecular reactions, making some modifications easier to accomplish, and (6) compatibility with automated synthesis technology.
Delivery of polypeptides in multi-kilogram quantities within commercially competitive timelines is extremely challenging, especially when coupled with a desire to minimize environmental and economic impact. This case study explores how CPC Scientific’s advanced process improvements and efficiencies within solid-phase peptide synthesis (SPPS) have enabled multi-kilogram delivery of a pharmaceutically-relevant decapeptide within a challenging timescale, driving sustainable and cost-saving production for the client.
Our team has developed an innovative DMF recycling strategy that substantially reduces solvent consumption during solid-phase peptide synthesis. Minimizing use of DMF, a major environmental and cost contributor in peptide manufacturing, has improved process sustainability and cost efficiency. This method neatly demonstrates how targeted green chemistry practices can be successfully integrated into large-scale SPPS, supporting more environmentally responsible and economically viable peptide production.
In Part 1 of our Minimal Protection Group Strategies for SPPS, we discussed methods for eliminating sidechain protection on hydroxy-bearing amino acids such as serine, threonine, tyrosine, and hydroxyproline. By omitting t-butyl protection, we enhanced atom economy and avoided the use of hazardous solvents typically required to remove these protection groups. In Part 2, we present a new case study, expanding our approach to include the unprotected side chains of histidine, tryptophan, and arginine. We demonstrate the synthesis of a Goserelin peptide API impurity, showcasing how a convergent peptide fragment strategy can be used to eliminate the need for TFA and diethyl ether, eliminate side chain protection of Arginine, Histidine, and Tryptophan.
The synthesis of the linear RP-182 analog, bicyclo[6.1.0]non-4-yn-9-ylmethyloxycarbonyl-PEG2-Lys-Phe-Arg-Lys-Ala-Phe-Lys-Arg-Phe-Phe-Lys(azido-PEG)-NH2, was achieved using standard solid-phase peptide synthesis (SPPS) protocols. After cleaving the linear peptide from the resin, macrocyclization was performed in the liquid phase through a strain-promoted click reaction. BCN introduces extra ring strain due to its fused cyclopropane structure. The combined effect of ring strain, the selection of BCN, and copper catalysis significantly increases the macrocyclization efficiency of longer peptides like RP-182.
Stapled peptides have emerged as a powerful tool in drug discovery and therapeutic development due to their ability to overcome the limitations associated with traditional peptide drugs, such as poor stability and low cell permeability. By introducing staples into the peptide backbone, researchers can stabilize peptide conformations and enhance their interactions with target proteins, resulting in improved efficacy and specificity. This approach not only addresses the challenges of peptide drug design but also opens new avenues for targeting challenging biomolecular interactions that are difficult to modulate with small molecules or antibodies. The development of stapled peptides has led to significant advancements in targeting protein-protein interactions, addressing previously intractable diseases, and enhancing the precision of therapeutic interventions.
Peptide receptor radionuclide therapy (PRRT) is a targeted therapeutic approach that utilizes peptides to deliver cytotoxic radiation to specific receptors overexpressed on cancer cells. Peptides offer several advantages as therapeutic vectors in PRRT due to their small size, favorable pharmacokinetics, high binding affinity, low immunogenicity and toxicity, and minimal off-target effects. Tumor-targeting peptides conjugated to radionuclide chelates represent a promising class of cancer therapeutics.
Most proteinogenic peptides, except for certain hydrophobic sequences, can be synthesized in a linear fashion using solid-phase peptide synthesis (SPPS) methods. However, longer sequences, particularly those exceeding 70 amino acids, often require alternative techniques due to challenges like steric hindrance. Other issues, such as poor solvation of the protected peptide during synthesis and the formation of intermolecular hydrogen bonds (e.g., β-sheets) between fragments, can also result in inefficient coupling and deprotection.
Synthetic oligonucleotides constitute an important class of therapeutics developed to treat a variety of indications. Two main synthetic approaches exist for the conjugation of a peptide to an oligonucleotide: parallel and linear. The primary benefit of the linear approach is the one-pot solid-phase assembly and compatibility with machine automation. However, in cases where poor compatibility of peptide and oligo chemistries exist or long peptide and oligo fragments are required, preparing both components separately and linking both compounds together may offer the simplest solution.
Solid-phase peptide synthesis (SPPS) approaches require that the side chains of certain amino acids be protected from undesired reactivity during synthesis. The installation and removal of these protection groups results in a lower atom economy in the production process. Removal of the protection groups often requires large volumes of trifluoroacetic acid (TFA) or other strong acids which can result in lower yields and pose a significant risk to the environment.
The transferred energy from a fluorescent donor is converted into molecular vibrations if the acceptor is a non-fluorescent dye (quencher). When the FRET is terminated (by separating donor and acceptor), an increase of donor fluorescence can be detected. The design and synthesis work at CPC for FRET and TR-FRET peptide substrates include modification of sequences, selection of donor/quencher pairs, improvement of FRET substrate solubility and quenching efficiency.
Genomics research shows that more than a million proteins are encoded by approximately 30,000 human genes. Proteomics, the study of proteins encoded by the genome, includes identifying post-translational modifications, structural analyses, protein localization studies, and protein quantitation. Mass spectroscopybased techniques have evolved as a powerful tool in proteomics. Stable isotope-labeled peptides (SIL peptides) are chemically and physically indistinguishable from their endogenous counterparts concerning retention time, ionization efficiency, and fragmentation pathways.
Peptides play a vital role in the pharmaceutical industry and drug therapeutic development; however, their in vivo applications are sometimes limited due to fast degradation by proteases, poor solubility, antigenic responses, and glomerular filtration in the kidney. The covalent attachment of polyethylene glycol (PEG) chains to peptides is one approach that can reduce immunogenicity, improve solubility, and reduce renal clearance.
Hydrocarbon-stapled peptides are locked into their bioactive alpha-helical conformation through the site-specific introduction of a chemical brace, an all-hydrocarbon staple. The idea of peptide stapling was introduced to overcome the limitations of two broad classes of therapeutic agents (small molecules and protein biologics) in targeting intracellular protein-protein interactions. Small molecules only work on proteins with a specific surface feature, and most protein biologics do not penetrate cells. Because stapled peptides are locked into a stabilized α-helical structure (the most common element of protein secondary structures), they can easily penetrate cells.
Protein-protein and protein-peptide interactions play critical roles in all types of cellular processing. Peptides are natural partners to proteins and, as ligands, bind to proteins with high affinity due to their capacity to adapt to the often flexible protein surface. Despite this, peptides have drawbacks as drug candidates that include low plasma bioavailability, instability from proteolytic enzymes, and poor passive membrane permeability. Some success has been achieved with linear peptides, particularly peptides that maintain α-helical secondary structures. These motifs can be introduced to stabilized α-helical motifs by common “peptide-stapling” approaches, but stapled peptides can suffer from low bioactivity and poor solubility. Another strategy to maintain peptide secondary structure is modification by macrocyclization.
Mutations to the RBD of SARS-CoV have resulted in the reorganization of SARS-CoV-2 RBD, specifically to the loops that are in close proximity to the ACE2-binding ridge. Due to these structural changes, an additional intramolecular main-chain hydrogen bond is gained between Asn487 and Ala475 (Figure 4a). The Asn487–Ala475 hydrogen bond causes the RBM of SARS-CoV-2 to be more structurally compact, enabling the loop containing Ala475 to move into closer proximity to the ACE2 α1 helix. Closer contact between the RBM and α1 helix of ACE2 results in more intermolecular interactions (Figure 4a,b). Some of these interactions include hydrogen bonds between Gln493 (SARS-CoV-2) and Glu35 (ACE2),[3b,c] and main-chain hydrogen bonds between Gly502 and Lys31.
This guide will help you understand how to read ESI spectra for large and complex bio-molecules like peptides. Other techniques such as high-performance liquid chromatography (HPLC) show one major peak (i.e., absorbance and purity for RP-HPLC) in the spectra, making the interpretation straightforward. ESI-MS spectra are more complicated due to the multiplicity of the data. While most small molecules (100-500 AMU) behave well in ESI-MS and ionize as a single charged state, larger biomolecules, such as peptides, ionize as multiple charged variants. A major advantage of ESI-MS over other mass spectroscopy techniques is that molecular fragmentation is rare (t-Boc groups and a few other protection groups are an exception), which is why ESI-MS is often referred to as a soft ionization technique.
Solid-phase peptide synthesis (SPPS) has many advantages over liquid-phase peptide synthesis (LPPS) for preparing and manufacturing synthetic peptides. Except the synthesis of short peptide sequences (i.e., less than five amino acid residues), SPPS is faster, more efficient, and more economical than liquid-phase peptide synthesis (LPPS). Some of the advantages of SPPS include: (1) Excess reagents and products can be easily washed away, (2) using excess reagents to increase reaction rates and drive reactions to completion, (3) intermediates do not require isolation or characterization, (4) access to a broader range of solvents with low volatility and high polarity, (5) tethered peptide provides a ‘pseudo-dilute’ microenvironment, which can inhibit intermolecular reactions, making some modifications easier to accomplish, and (6) compatibility with automated synthesis technology.